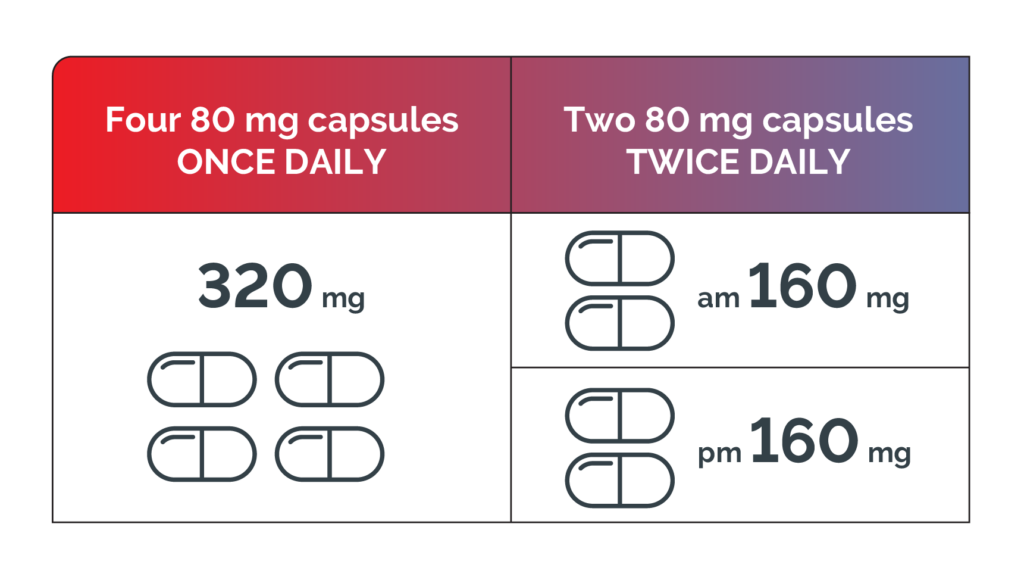
BRUKINSA®▼ is an oral monotherapy with a flexible dosing regimen, providing a convenient chemotherapy-free treatment.1 The recommended dosing for BRUKINSA is the same for all approved indications.1
Brukinsa dosing flexibility1
BRUKINSA is for oral use. The hard capsules can be taken with or without food. Patients should be instructed to swallow the capsules whole with water, and not to open, break or chew the capsules.1
The recommended total daily dose of BRUKINSA is 320 mg. The daily dose may be taken either once daily (four 80 mg capsules) or divided into two doses of 160 mg twice daily (two 80 mg capsules). Treatment with BRUKINSA should be continued until disease progression or unacceptable toxicity.1
No clinically significant differences in BRUKINSA pharmacokinetics were observed when co-administered with gastric acid reducing agents (proton pump inhibitors, H2-receptor antagonists).1
Recommended dose adjustment for
≥Grade 3 adverse reactions1
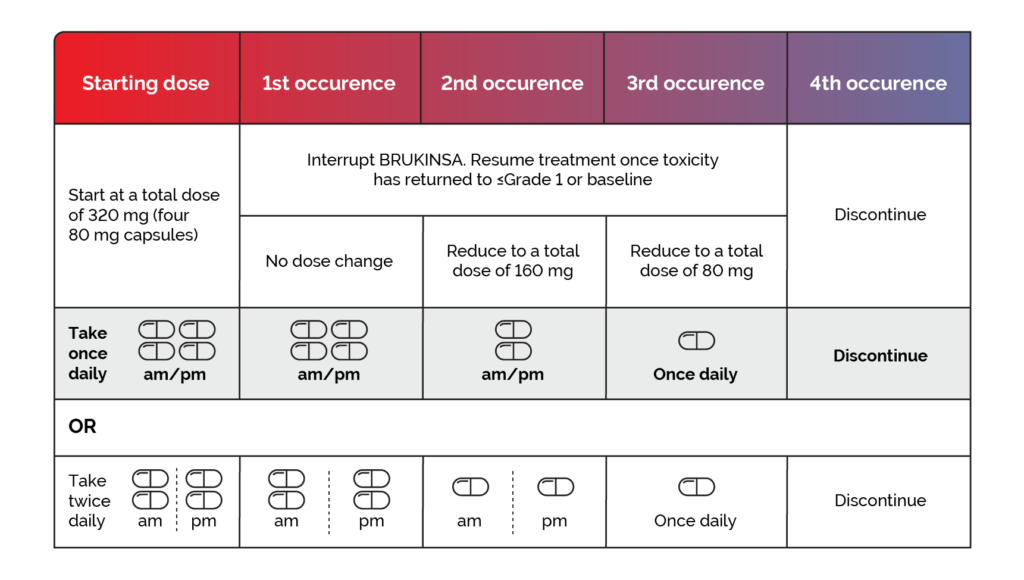
Dose adjustments are recommended when adverse reactions occur at Grade 3 or above. Please refer to the BRUKINSA Summary of Product Characteristics for further information.1
Interrupt BRUKINSA therapy after the first, second or third adverse reaction occurrence and resume at the doses provided in Figure 1 once toxicity has resolved to ≤Grade 1 or baseline. After the fourth adverse reaction occurrence, discontinue BRUKINSA.1
Recommended dose modifications of BRUKINSA for Grade 3 or greater adverse reactions are provided in Figure 1, and include:1
- ≥Grade 3 non-haematological toxicities
- Grade 3 febrile neutropenia
- Grade 3 thrombocytopenia with significant bleeding
- Grade 4 neutropenia (lasting >10 consecutive days)
- Grade 4 thrombocytopenia (lasting >10 consecutive days)
Asymptomatic lymphocytosis should not be regarded as an adverse reaction, and these patients should continue taking BRUKINSA.1
Figure 1. Recommended BRUKINSA dose modifications for ≥ Grade 3 adverse reactions.1*
Recommended dose modifications for use with CYP3A inhibitors or inducers1
Recommended dose modifications for the use of BRUKINSA with CYP3A inhibitors or inducers are provided in Table 1.
Table 1. Recommended BRUKINSA dose modifications when co-administered with other medicinal products.1
| CYP3A | Co-administered medicinal product | Recommended dose |
|---|---|---|
| Inhibition | Strong CYP3A inhibitor (e.g., posaconazole, voriconazole, ketoconazole, itraconazole, clarithromycin, indinavir, lopinavir, ritonavir, telaprevir) Moderate CYP3A inhibitor (e.g., erythromycin, ciprofloxacin, diltiazem, dronedarone, fluconazole, verapamil, aprepitant, imatinib, grapefruit juice, Seville oranges) | 80 mg once daily 80 mg twice daily |
| Induction | Strong CYP3A inducer (e.g., carbamazepine, phenytoin, rifampin, St. John’s wort) Moderate CYP3A inducer (e.g., bosentan, efavirenz, etravirine, modafinil, nafcillin) | Avoid concomitant use; consider alternative agents with less CYP3A induction |
Missed dose1
A double dose should not be taken to make up for a forgotten dose. If a dose is not taken at the scheduled time, the next dose should be taken according to the normal schedule.
Special populations1
Elderly
No specific dose adjustment is required for elderly patients (aged ≥65 years).
Renal impairment
No dose modification is recommended in patients with mild to moderate renal impairment (creatinine clearance (CrCl) ≥30 mL/min, estimated by Cockcroft-Gault). There is limited data on patients with severe renal impairment and end-stage renal disease (n=12). Patients with severe renal impairment (CrCl <30 mL/min) or on dialysis should be monitored for adverse reactions.
Hepatic impairment
Dose modifications are not needed in patients with mild (Child-Pugh class A) or moderate hepatic impairment (Child-Pugh class B). Patients with mild or moderate hepatic impairment were treated in BRUKINSA clinical studies. The recommended dose of BRUKINSA for patients with severe hepatic impairment (Child-Pugh class C) is 80 mg orally twice daily. The safety of BRUKINSA has not been evaluated in patients with severe hepatic impairment. Monitor these patients closely for adverse events of BRUKINSA.
Paediatric population
The safety and efficacy of BRUKINSA in children and adolescents below 18 years of age have not been established. No data are available.
Please refer to the BRUKINSA Summary of Product Characteristics for further information.