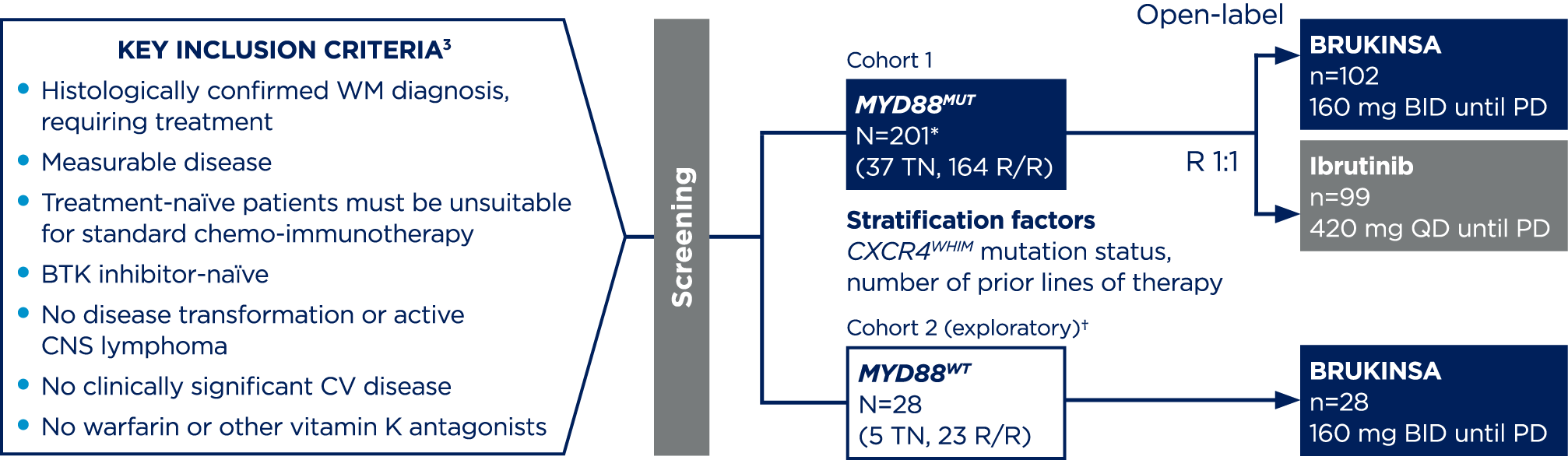
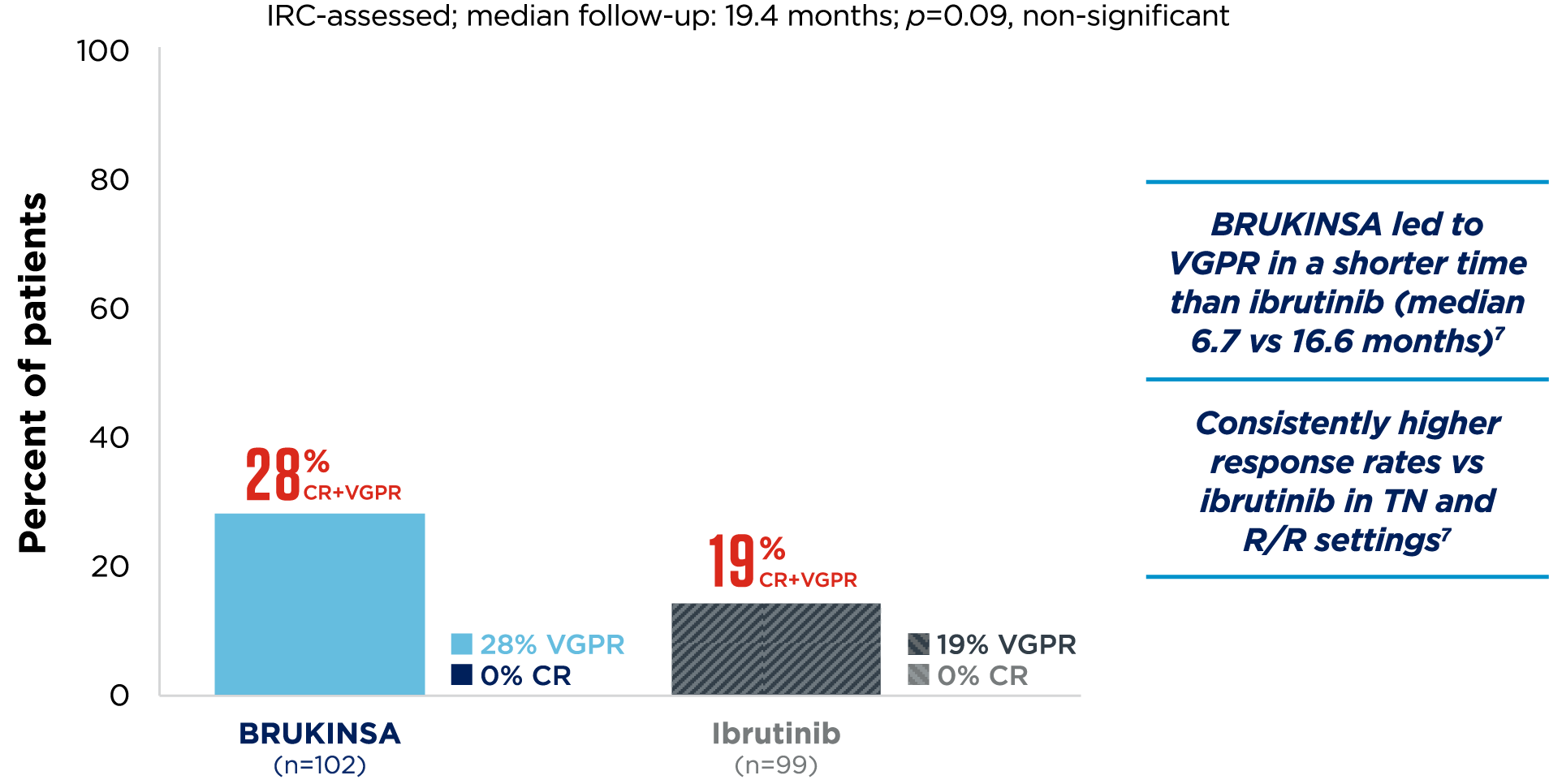
*201 patients were randomised, 199 received ≥1 dose of the study treatment.3
†Cohort 2 rationale: Since major responses have not previously been observed in ibrutinib-treated patients with MYD88WT, patients found to have MYD88WT by gene sequencing (n=26) or those with unknown/inconclusive MYD88WT mutational status (n=2) were assigned to receive BRUKINSA in this separate single-arm exploratory analysis.4
‡Overall safety data are from the larger study population: BRUKINSA n=101, ibrutinib n=98. At >36 months: BRUKINSA n=72, ibrutinib n=64.7
¶BGGB-3111-LTE1 is a long-term extension study enrolling patients treated with BRUKINSA from parent trials treating B-cell malignancies.8
AE, adverse event; BID, twice daily; BTK, Bruton’s tyrosine kinase; CI, confidence interval; CLL, chronic lymphocytic leukaemia; CNS, central nervous system; CR, complete response; CV, cardiovascular; DOR, duration of response; IRC, independent review committee; IWWM, International Workshop on Waldenström’s Macroglobulinaemia; MCL, mantle cell lymphoma; MRR, major response rate; MUT, mutation; MZL, marginal zone lymphoma; NE, non-evaluable; ORR, overall response rate; PD, progressive disease; PFS, progression-free survival; PR, partial response; QD, once daily; R, randomisation; R/R, relapsed/refractory; SD, stable disease; TEAE, treatment-emergent adverse event; TN, treatment naïve; VGPR(+), very good partial response (or better); WHIM, warts, hypogammaglobulinaemia infections, and myelokathexis; WM, Waldenström’s macroglobulinaemia; WT, wildtype.
References:
- BRUKINSA. United Kingdom Summary of Product Characteristics. BeOne Medicines UK Ltd.;
- Tam CS, et al. Blood Cancer J. 2023;13(1):141;
- Tam CS, et al. Blood. 2020;136(18):2038–2050;
- Dimopoulos M, et al. Blood Adv. 2020;4(23):6009–6018;
- Owen R, et al. Hemasphere. 2022;(6):1020–1021;
- Castillo JJ, et al. Leukemia. 2022;36:532–539;
- Dimopoulos MA, et al. J Clin Oncol. 2023;41(33):5099–5106;
- D’Sa S, et al. ASH annual meeting. December 7–10, 2024. Poster 3031;
- Tam CS, et al. Expert Rev Clin Pharmacol. 2021;14(11):1329–1344;
- Brullo C, et al. Int J Mol Sci. 2021;22(14):7641;
- Shadman M, et al. Lancet Haematol. 2023;10(1):e35–e45;
- Guo Y, et al. J Med Chem. 2019;62(17):7923–7940;
- Imbruvica. Summary of Product Characteristics. Pharmacyclics LLC, Janssen Biotech, Inc.;
- Calquence. Summary of Product Characteristics. AstraZeneca Pharmaceuticals LP.
The study is registered with ClinicalTrials.gov: NCT03053440